11 Plot ORA
After selecting interested terms or pathways from genORA or genGSEA result, users can pass the data frame to plotEnrich, which includes many ready-made plot types, including barplot, dotplot, heatmap, wego-like plot, chord plot, network, wordcloud etc.
It is worth mentioning that almost all plots are based on ggplot2 and the plot_theme function can easily change their border, legend, label etc., which helps users make their own plots.
11.1 Get ORA result
For more details, please refer to chapter 8
# 1st step: prepare input IDs
data(geneList, package = "genekitr")
entrez_id <- names(geneList)[abs(geneList) > 2]
head(entrez_id, 5)## [1] "948" "1638" "158471" "10610" "6947"
# 2nd step: prepare gene set
hg_gs <- geneset::getGO(org = "human",ont = "bp")
# 3rd step: ORA analysis
ego <- genORA(entrez_id, geneset = hg_gs, p_cutoff = 0.01, q_cutoff = 0.01)
# next we only show ten sample terms
ego <- ego[1:10, ]
head(ego)## Hs_BP_ID
## GO:1901992 GO:1901992
## GO:0050673 GO:0050673
## GO:1901989 GO:1901989
## GO:0048660 GO:0048660
## GO:0048659 GO:0048659
## GO:0006260 GO:0006260
## Description
## GO:1901992 positive regulation of mitotic cell cycle phase transition
## GO:0050673 epithelial cell proliferation
## GO:1901989 positive regulation of cell cycle phase transition
## GO:0048660 regulation of smooth muscle cell proliferation
## GO:0048659 smooth muscle cell proliferation
## GO:0006260 DNA replication
## GeneRatio BgRatio pvalue p.adjust qvalue
## GO:1901992 0.06451613 105/19160 4.044808e-07 0.0007297257 0.0005741369
## GO:0050673 0.12096774 482/19160 5.326465e-07 0.0007297257 0.0005741369
## GO:1901989 0.06451613 125/19160 1.532079e-06 0.0009559183 0.0007521017
## GO:0048660 0.07258065 174/19160 1.980715e-06 0.0009559183 0.0007521017
## GO:0048659 0.07258065 178/19160 2.389740e-06 0.0009559183 0.0007521017
## GO:0006260 0.08870968 284/19160 2.402775e-06 0.0009559183 0.0007521017
## geneID
## GO:1901992 407007/113130/6241/595/407006/993/9134/51514
## GO:0050673 5176/26471/3488/6591/8549/6657/4208/407007/6422/29128/595/7039/374/3690/10468
## GO:1901989 407007/113130/6241/595/407006/993/9134/51514
## GO:0048660 3488/3725/10891/4208/3248/407007/407006/7099/3690
## GO:0048659 3488/3725/10891/4208/3248/407007/407006/7099/3690
## GO:0006260 5427/157570/641/5888/8318/993/9156/9134/55388/51514/374393
## geneID_symbol
## GO:1901992 MIR222/CDCA5/RRM2/CCND1/MIR221/CDC25A/CCNE2/DTL
## GO:0050673 SERPINF1/NUPR1/IGFBP5/SNAI2/LGR5/SOX2/MEF2C/MIR222/SFRP1/UHRF1/CCND1/TGFA/AREG/ITGB3/FST
## GO:1901989 MIR222/CDCA5/RRM2/CCND1/MIR221/CDC25A/CCNE2/DTL
## GO:0048660 IGFBP5/JUN/PPARGC1A/MEF2C/HPGD/MIR222/MIR221/TLR4/ITGB3
## GO:0048659 IGFBP5/JUN/PPARGC1A/MEF2C/HPGD/MIR222/MIR221/TLR4/ITGB3
## GO:0006260 POLE2/ESCO2/BLM/RAD51/CDC45/CDC25A/EXO1/CCNE2/MCM10/DTL/FAM111B
## Count FoldEnrich RichFactor
## GO:1901992 8 11.772657 0.07619048
## GO:0050673 15 4.808593 0.03112033
## GO:1901989 8 9.889032 0.06400000
## GO:0048660 9 7.992214 0.05172414
## GO:0048659 9 7.812613 0.05056180
## GO:0006260 11 5.984780 0.0387323911.2 Bar Plot
Bar plot is the simplest way to show enriched terms. The x-axis is an enrichment metric (e.g. Gene ratio; Fold enrichment); the y-axis is selected terms. The bar color represents the statistical value.
What’s the difference between pvalue and p.adjust?
p.adjusthas seven adjustment methods: https://www.rdocumentation.org/packages/stats/versions/3.5.0/topics/p.adjust. The adjusted p-value is always the p-value, multiplied with some factor: adj.p = f * p. The actual size of this factor f depends on the strategy used to correct for multiple testing. By the way, the “q-value” stands for the “false discovery rate (FDR)” method.
The basic arguments are:
-
term_metric: The x-axis could be “Count”, “GeneRatio”, “FoldEnrich” or “RichFactor” -
stats_metric: Statistic value of “p.adjust”, “pvalue” or “qvalue” -
up_color: Color for stronger statistic value (e.g. pvalue 0.01) -
down_color: Color for weaker statistic value (e.g. pvalue 1) -
wrap_length: Wrap term text if longer than this number
library(patchwork)
p1 <- plotEnrich(ego, plot_type = "bar")
p2 <- plotEnrich(ego, plot_type = "bar", term_metric = "GeneRatio", stats_metric = "pvalue")
p3 <- plotEnrich(ego, plot_type = "bar", up_color = "#E69056", down_color = "#325CAC")
p4 <- plotEnrich(ego, plot_type = "bar", wrap_length = 25)
p1 + p2 + p3 + p4 + plot_annotation(tag_levels = "A")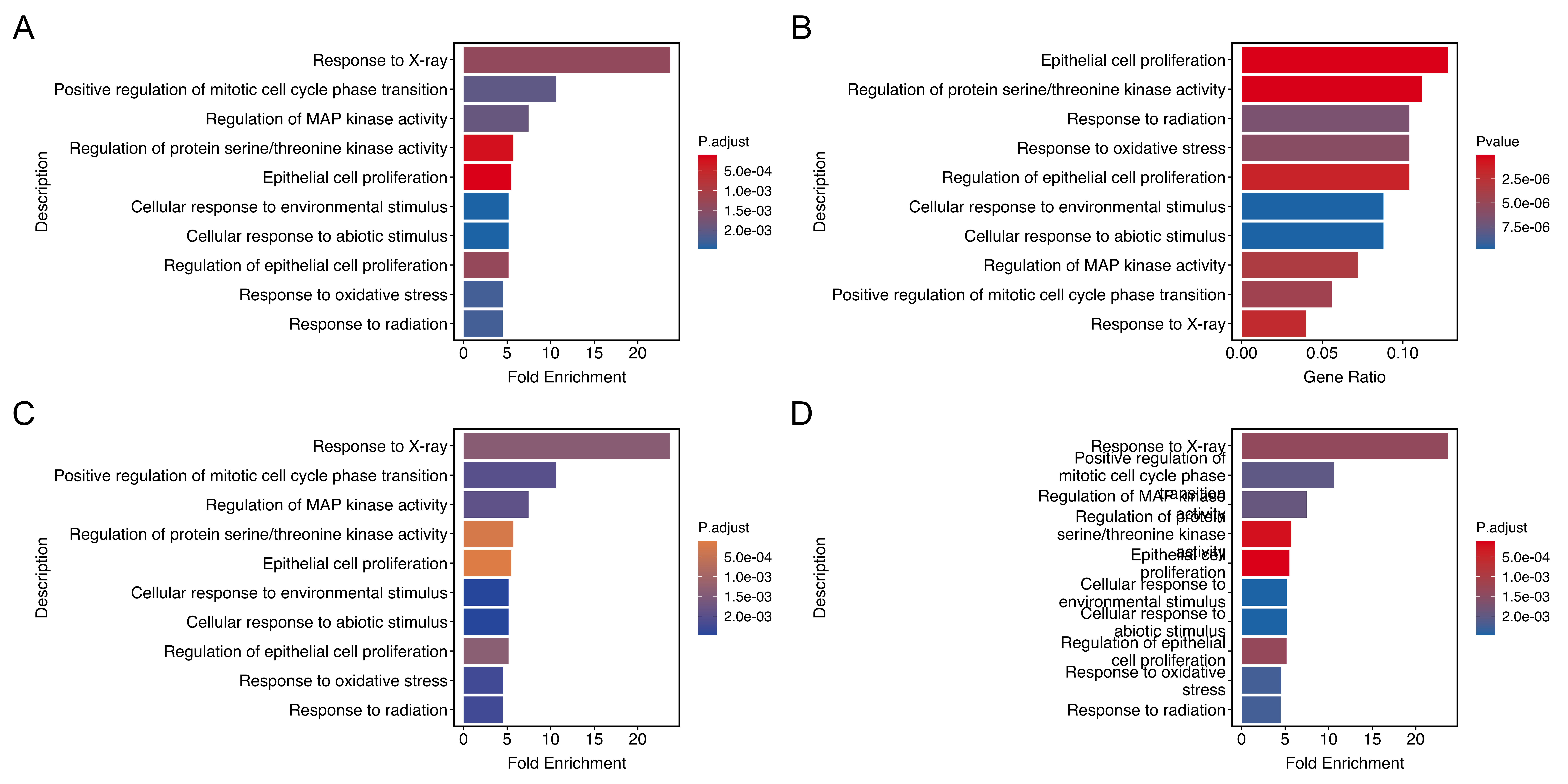
Figure 11.1: Bar plot of enrichment analysis. default (A), modify metrics (B), modify color (C) and modify term length (D).
11.3 Bubble Plot
Enriched gene sets are shown as bubbles with different sizes.
The x-axis is the statistic value and the y-axis is “Fold Enrichment”.
The basic arguments are:
-
stats_metric: Statistic value of “p.adjust”, “pvalue” or “qvalue” -
scale_ratio: Change bubble size. Default is 1.
library(patchwork)
p1 <- plotEnrich(ego, plot_type = "bubble")
p2 <- plotEnrich(ego, plot_type = "bubble",
scale_ratio = 0.5, stats_metric = "qvalue")
p1 / p2 + plot_annotation(tag_levels = "A")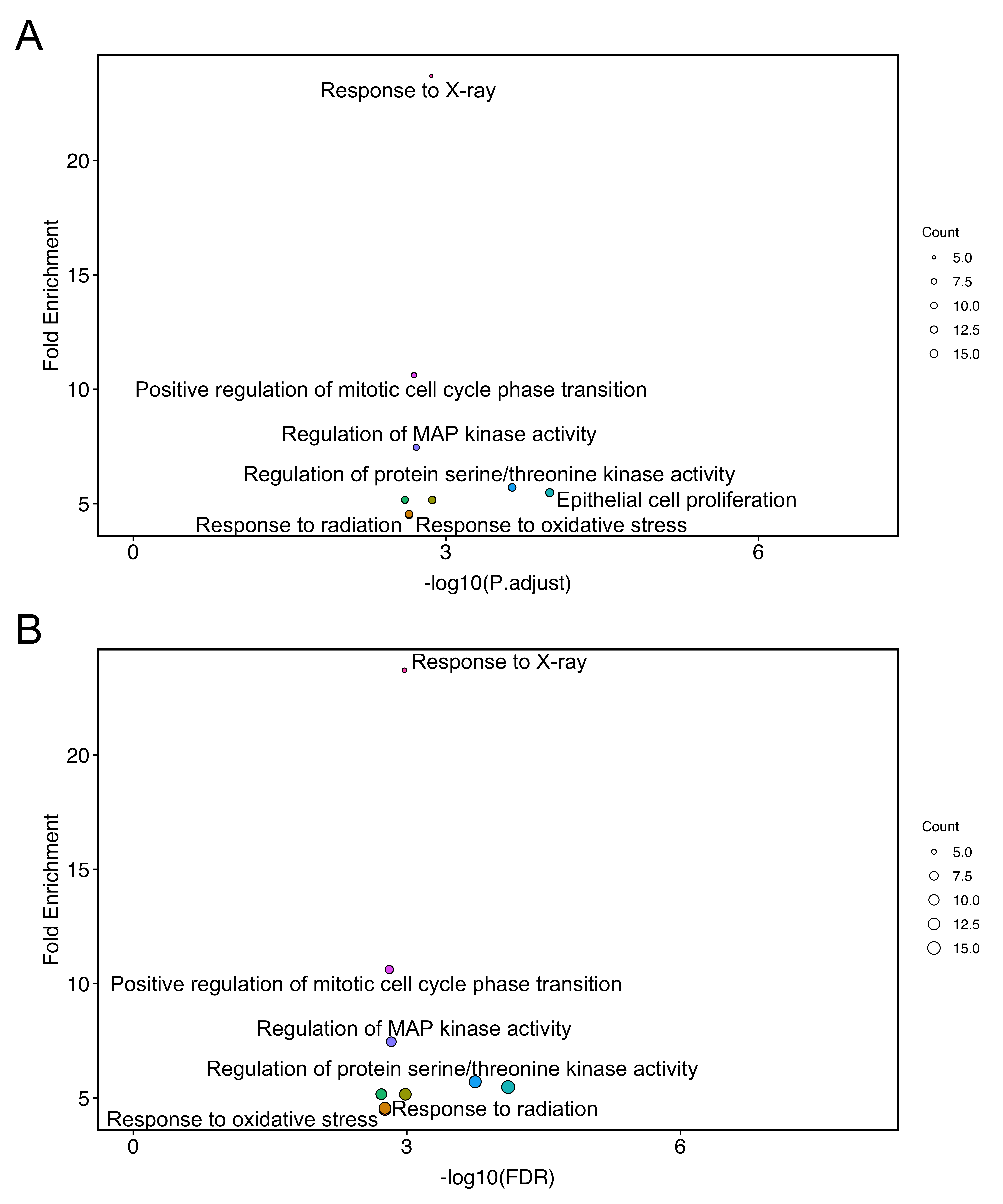
Figure 11.2: Bubble plot of enrichment analysis. default (A), modify bubble size (B).
11.4 Dot Plot
Similar to the bar plot, the dot plot is also widely used in enrichment analysis plotting. Like the bubble plot, dot size represents the gene number of an enriched term.
library(patchwork)
p1 <- plotEnrich(ego, plot_type = "dot")
p2 <- plotEnrich(ego,
plot_type = "dot",
scale_ratio = 1.5,
stats_metric = "pvalue",
term_metric = "RichFactor"
)
p1 + p2 + plot_annotation(tag_levels = "A")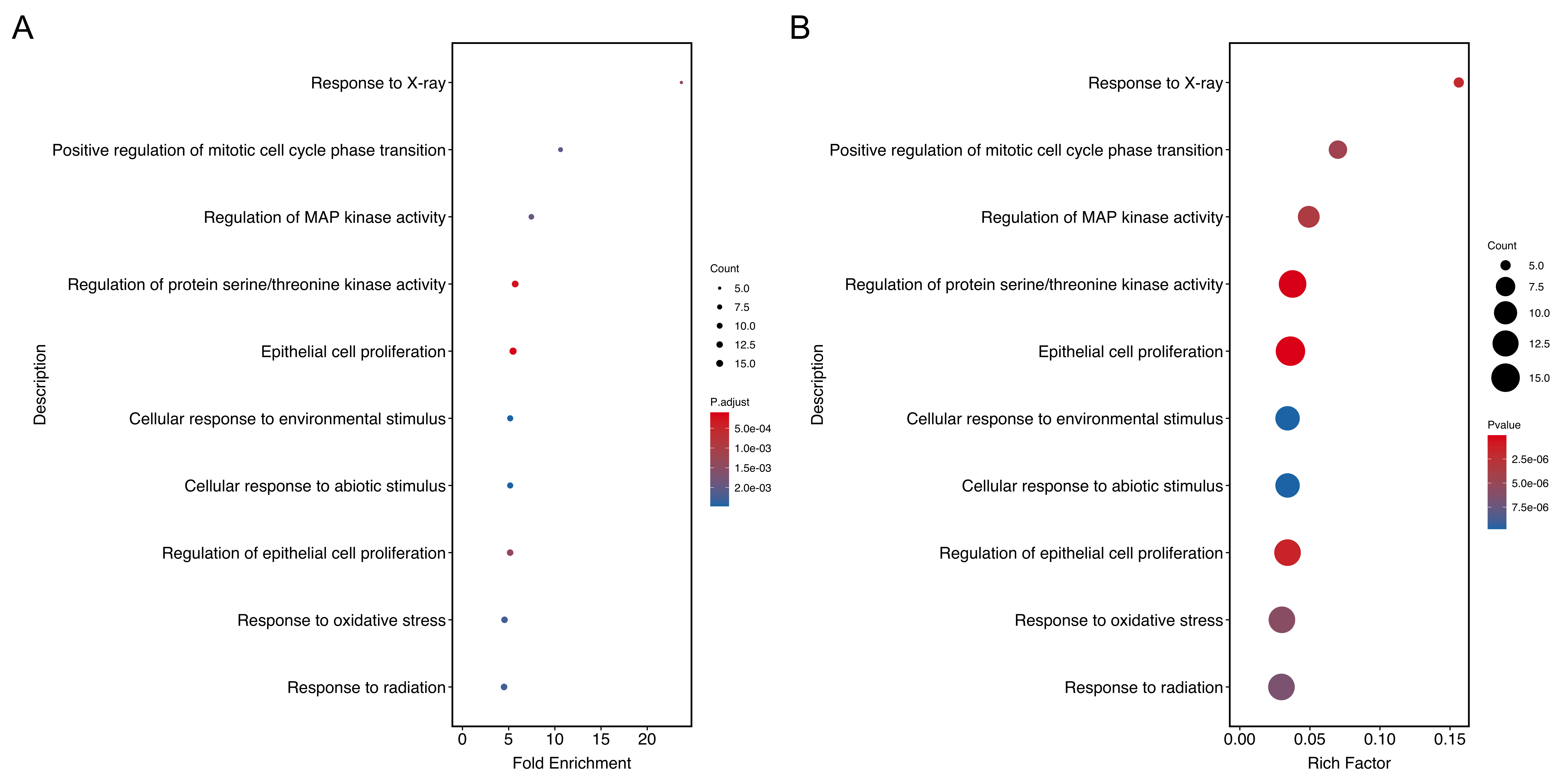
Figure 11.3: Dot plot of enrichment analysis. default (A), modify bubble size (B).
After analyzing group enrichment analysis for ORA, we can use dotplot to show the result:
plotEnrich(gego2,
plot_type = 'dot',
scale_ratio = 2, # dot size
main_text_size = 10,
legend_text_size =8,
n_term = 6) # show terms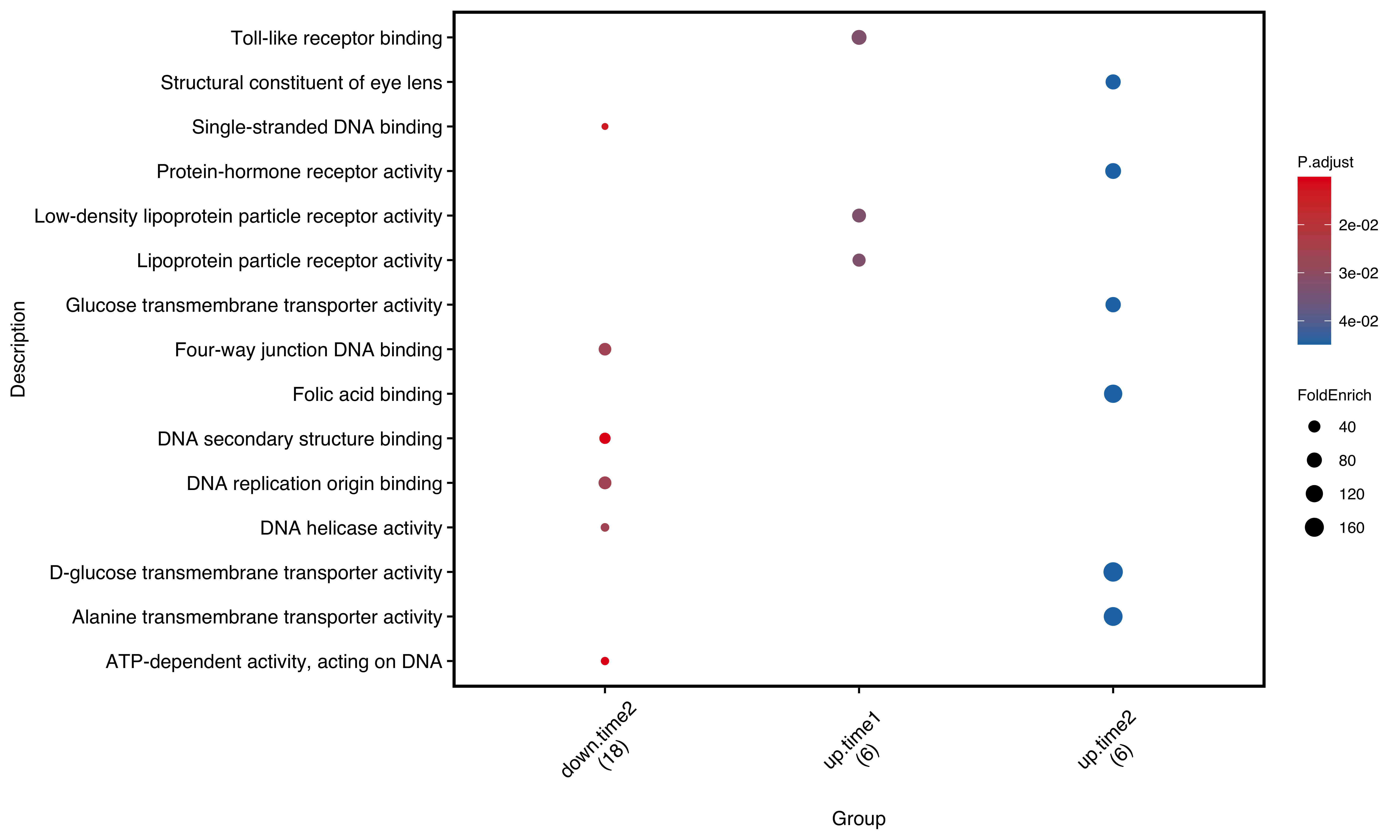
Figure 11.4: Dot plot of group enrichment analysis. number in round brackets shows total gene number in selected pathways
11.5 Lollipop Plot
The lollipop plot is like a combination of the barplot and the dotplot.
library(patchwork)
p1 <- plotEnrich(ego, plot_type = "lollipop")
p2 <- plotEnrich(ego,
plot_type = "lollipop",
scale_ratio = 1.5,
stats_metric = "pvalue",
term_metric = "RichFactor",
up_color = "#a32a31",
down_color = "#f7dcca"
)
p1 + p2 + plot_annotation(tag_levels = "A")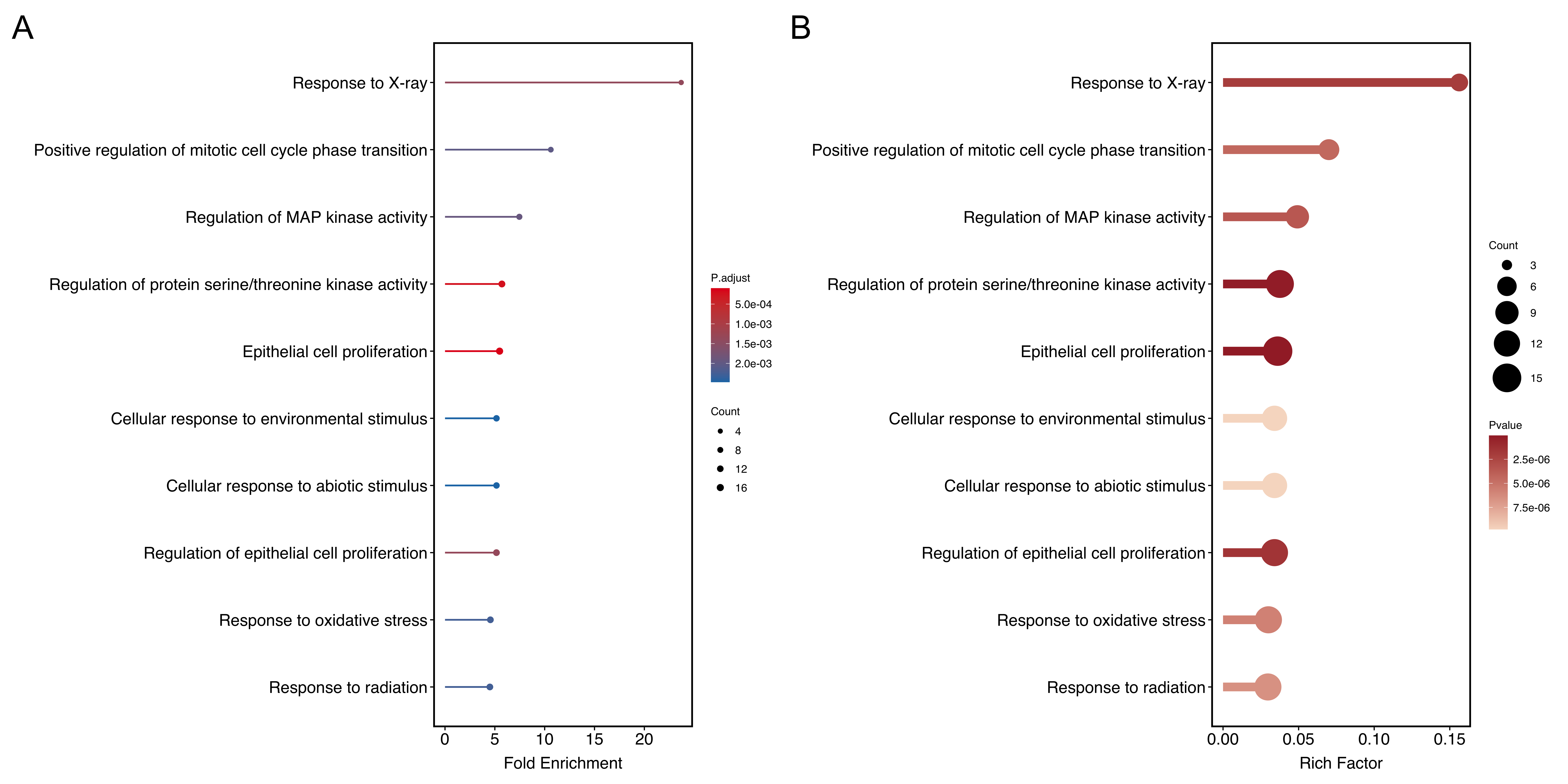
Figure 11.5: Lollipop plot of enrichment analysis. default (A), selected genes (B)
11.6 Heatmap Plot
The heatmap plot shows interactions between enriched terms and their genes. If fold change is given, the heatmap will add color for up- and down-regulated genes.
library(patchwork)
logfc <- geneList
p1 <- plotEnrich(ego, plot_type = "geneheat")
show_gene = c('JUN','SOX2','CD24','TLR4')
p2 <- plotEnrich(ego, plot_type = "geneheat", show_gene = show_gene)
p3 <- plotEnrich(ego, plot_type = "geneheat", show_gene = show_gene, fold_change = logfc)
p1 / p2 / p3 + plot_annotation(tag_levels = "A")
Figure 11.6: Heatmap plot of enrichment analysis. all genes (A, default), selected genes (B), selected genes with logFC value (C).
11.7 Chord Plot
Inspired by GOplot, the chord plot is reproduced using ggplot2 and it shows content similar to the heatmap plot.
-
gene_space: The space between the gene labels and the chord.
library(patchwork)
logfc <- geneList
show_gene = c('JUN','SOX2','CD24','TLR4')
p1 <- plotEnrich(ego, plot_type = "genechord",
show_gene = show_gene) +
ggplot2::theme(legend.position = "none")
p2 <- plotEnrich(ego, plot_type = "genechord",
show_gene = show_gene,
fold_change = logfc,
remove_legend_text = T,
gene_space = 0.5)
p1 + p2 + plot_annotation(tag_levels = "A")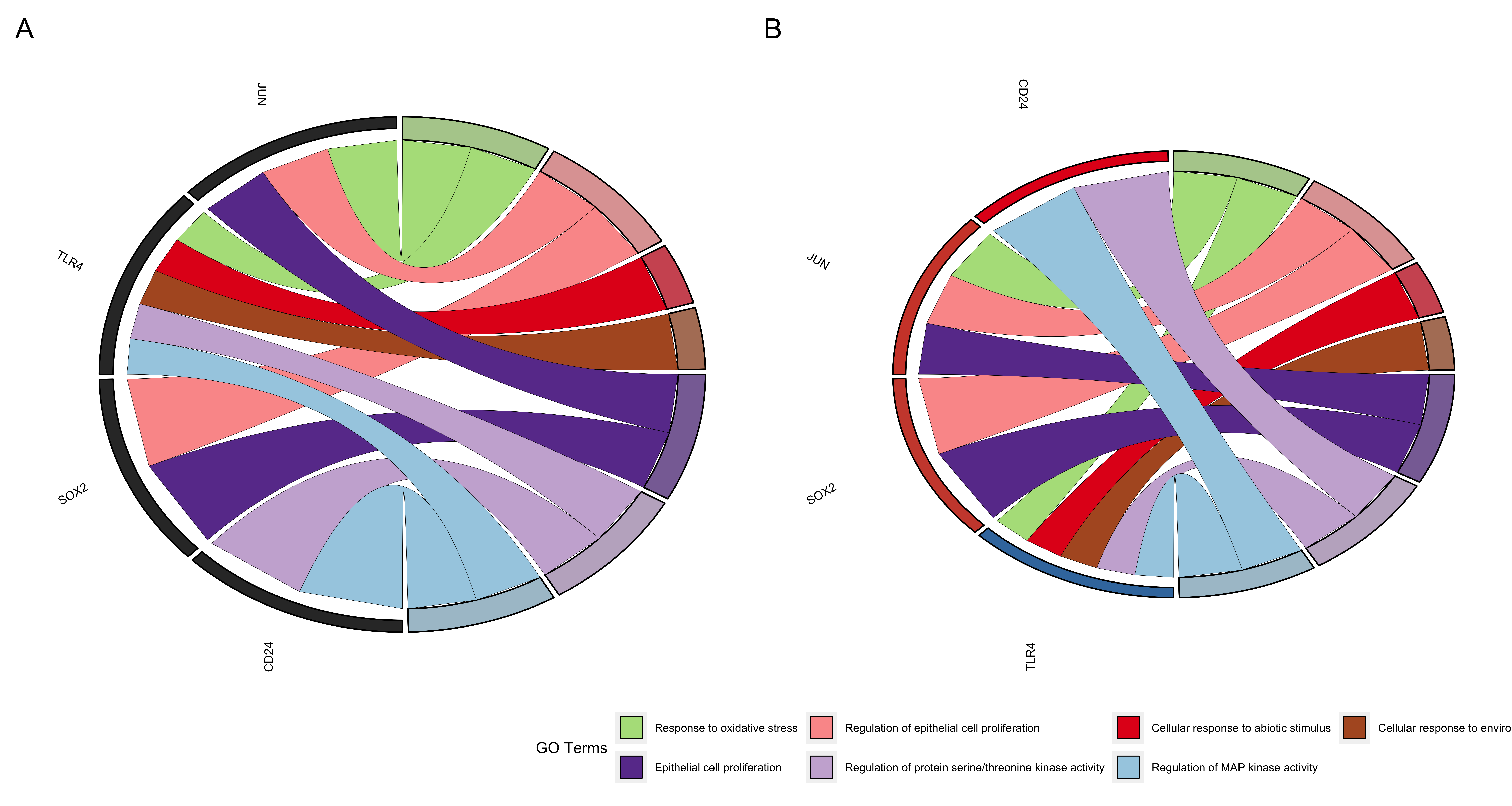
Figure 11.7: Chord plot of enrichment analysis. selected genes (A), selected genes with logFC value (B).
11.8 Wordcloud Plot
The wordcloud plot shows term emphasis based on text frequency.
plotEnrich(ego, plot_type = "wordcloud")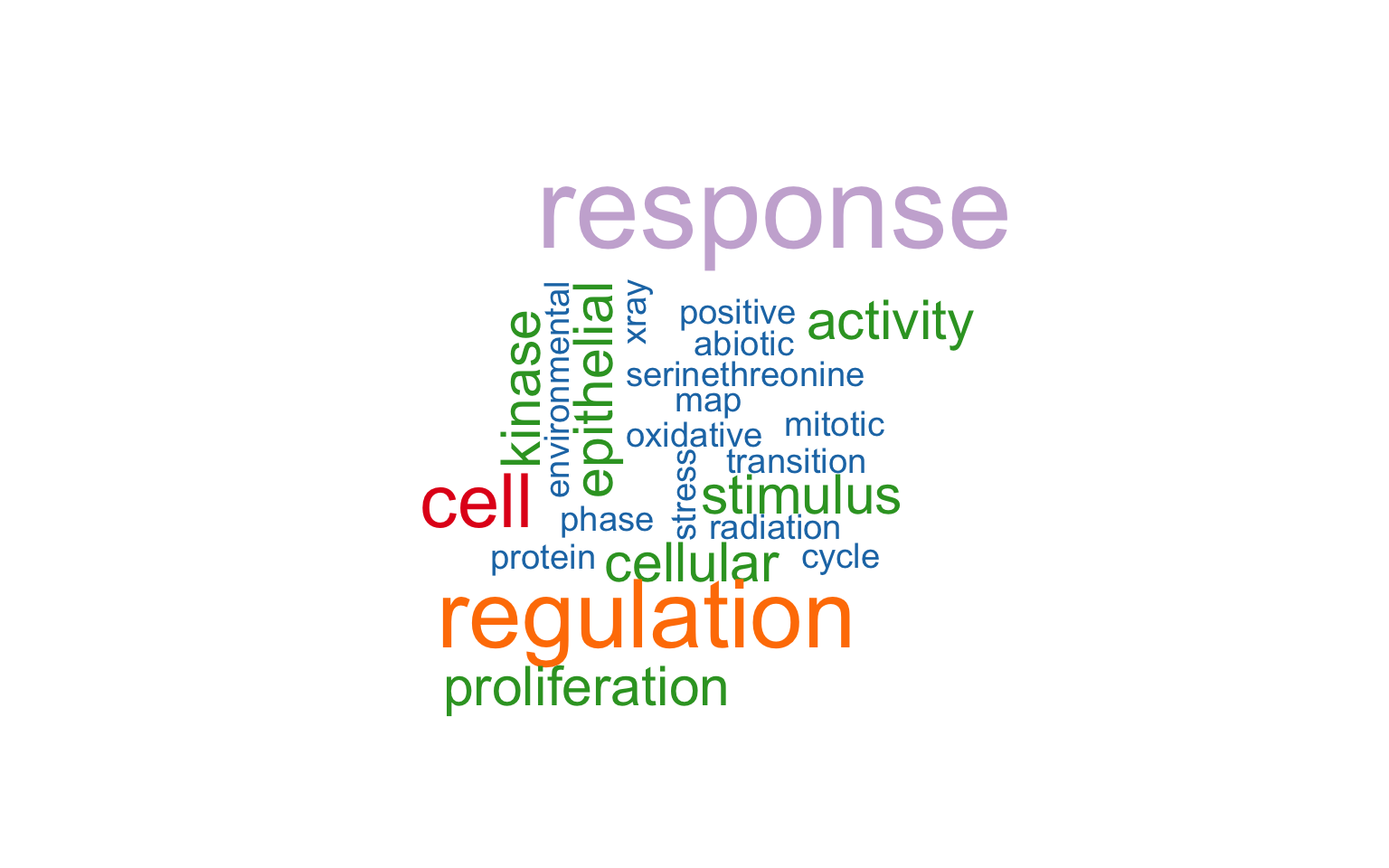
Figure 11.8: Wordcloud plot of enrichment analysis.
11.9 Upset Plot
Inspired by ComplexUpset, the upset plot shows the association between genes and enriched terms. Unlike a common Venn diagram, it can emphasize the complex relationships among many gene sets.
plotEnrich(ego, plot_type = "upset",main_text_size = 15,legend_text_size = 8)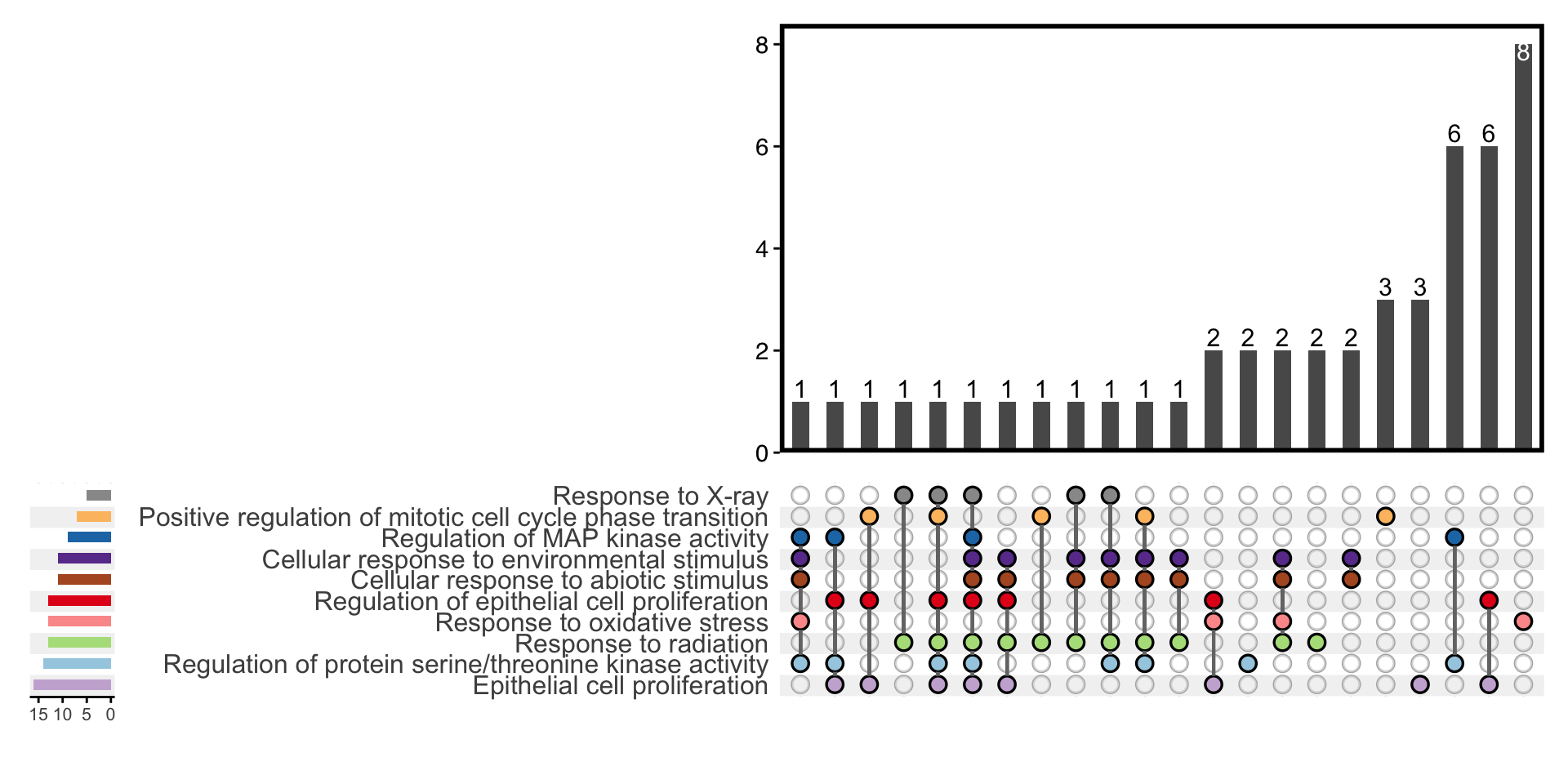
Figure 11.9: Upset plot of enrichment analysis.
11.10 Network Plot
Inspired by enrichplot::emapplot, genekitr reproduced this plot to enhance customization.
Enriched terms are the nodes in the network, and the overlapping gene sets are edges. Terms with more overlapping genes tend to cluster together, and the edges will be thicker.
- GO analysis results can use five semantic similarity methods to calculate the similarity between nodes: “Resnik”, “Lin”, “Rel”, “Jiang”, “Wang”, as well as the “JC” (Jaccard
- KEGG only supports the “JC” method.
Users can define the layout argument derived from ggraph, including “nicely” (default), “circle”, “dh”, “drl”, “fr”, “graphopt”, “grid”,“lgl”, “kk”, “mds”, “randomly”, “star” etc.
For more information about the
layout, you could refer to: “Introduction to ggraph: Layouts”
library(patchwork)
library(igraph)
library(ggraph)
p1 <- plotEnrich(ego, plot_type = "network", scale_ratio = 0.5)
p2 <- plotEnrich(ego, plot_type = "network",
layout = "circle", scale_ratio = 0.5)
p3 <- plotEnrich(ego, plot_type = "network",
layout = "grid", sim_method = "Wang",
up_color = "#a32a31", down_color = "#f7dcca")
(p1 + p2) / p3 + plot_annotation(tag_levels = "A")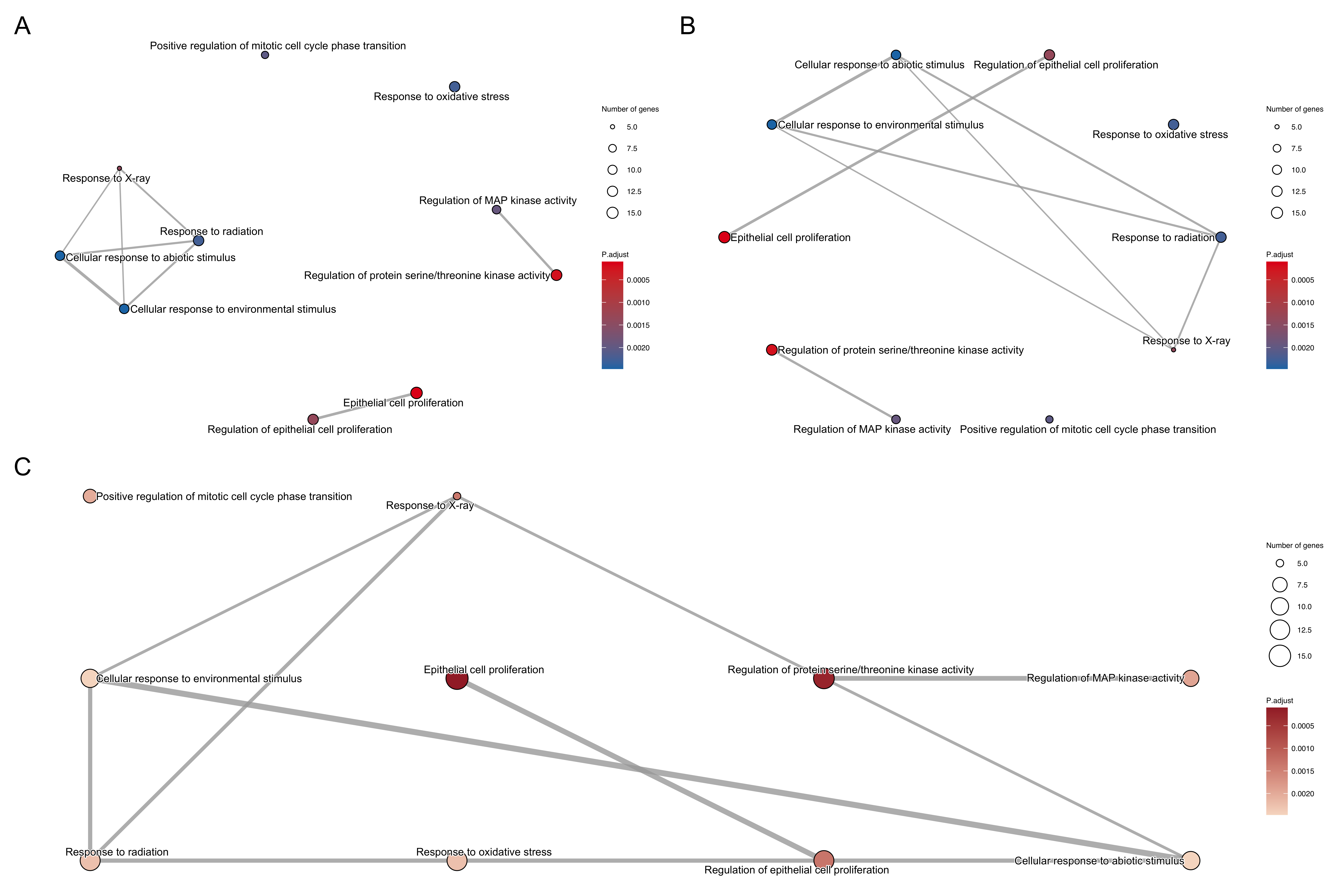
Figure 11.10: Network plot of enrichment analysis. JC method and nicely layout (A, default), circle layout (B), grid layout and Wang method (C).
11.11 GO-specific: WEGO Plot
To visualize more than one ontology of GO in one plot, users can use the
wego plot
Inspired by WEGO, genekitr utilized ggplot2 to reproduce this plot.
Here we generate two ontologies (MF and CC) result.
# 1st step: prepare input IDs
data(geneList, package = "genekitr")
id <- names(geneList)[abs(geneList) > 2]
# 2nd step: prepare CC and MF gene sets
go_cc <- geneset::getGO(org = "human",ont = "cc")
go_mf <- geneset::getGO(org = "human",ont = "mf")
# 3rd step: analysis
ego_cc <- genORA(id, geneset = go_cc)
ego_mf <- genORA(id, geneset = go_mf)
# 4th step: merge two data frames
# Note: each data frame should add new column "Ontology"
ego_cc <- ego_cc %>% dplyr::mutate(Ontology = "cc") %>% dplyr::rename(ID = 1)
ego_mf <- ego_mf %>% dplyr::mutate(Ontology = "mf") %>% dplyr::rename(ID = 1)
all_ego <- rbind(ego_cc,ego_mf)NOTICE: plotEnrich has a parameter n_term for the WEGO plot, which specifies the number of terms. If you want to plot all terms, just set n_term higher.
plotEnrich(all_ego, plot_type = "wego", n_term = 5)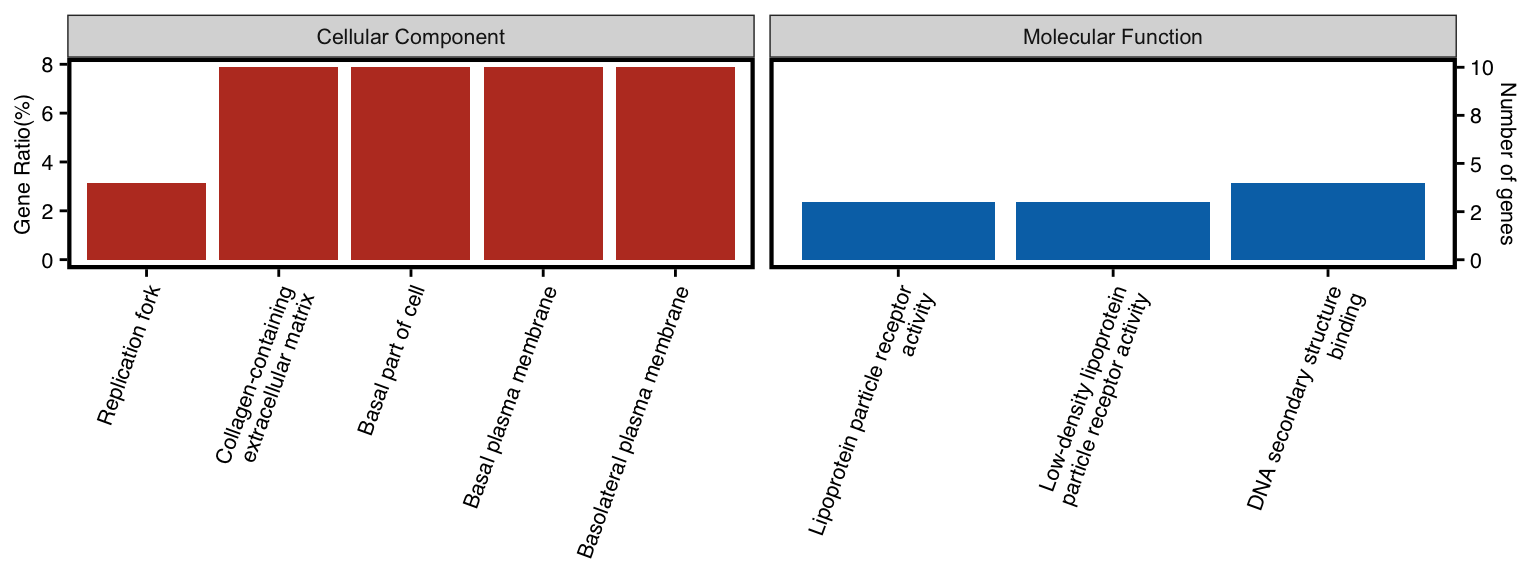
Figure 11.11: WEGO plot of enrichment analysis.
11.12 GO-specific: Map Plot
GO terms are built in a directed acyclic graph with a parent-child relationship. Here the map plot utilized GOSemSim to extract parent and child terms, and also utilized ggraph and igraph to draw with the default layout “sugiyama”.
To avoid cluttering the plot with too many unrelated terms, genekitr only shows the closest parent and child of the selected terms. Besides, the top three parent terms with more edges will be plotted.
library(igraph)
library(ggraph)
plotEnrich(ego, plot_type = "gomap", wrap_length = 25,
up_color = '#a32a31',down_color = '#3665a6')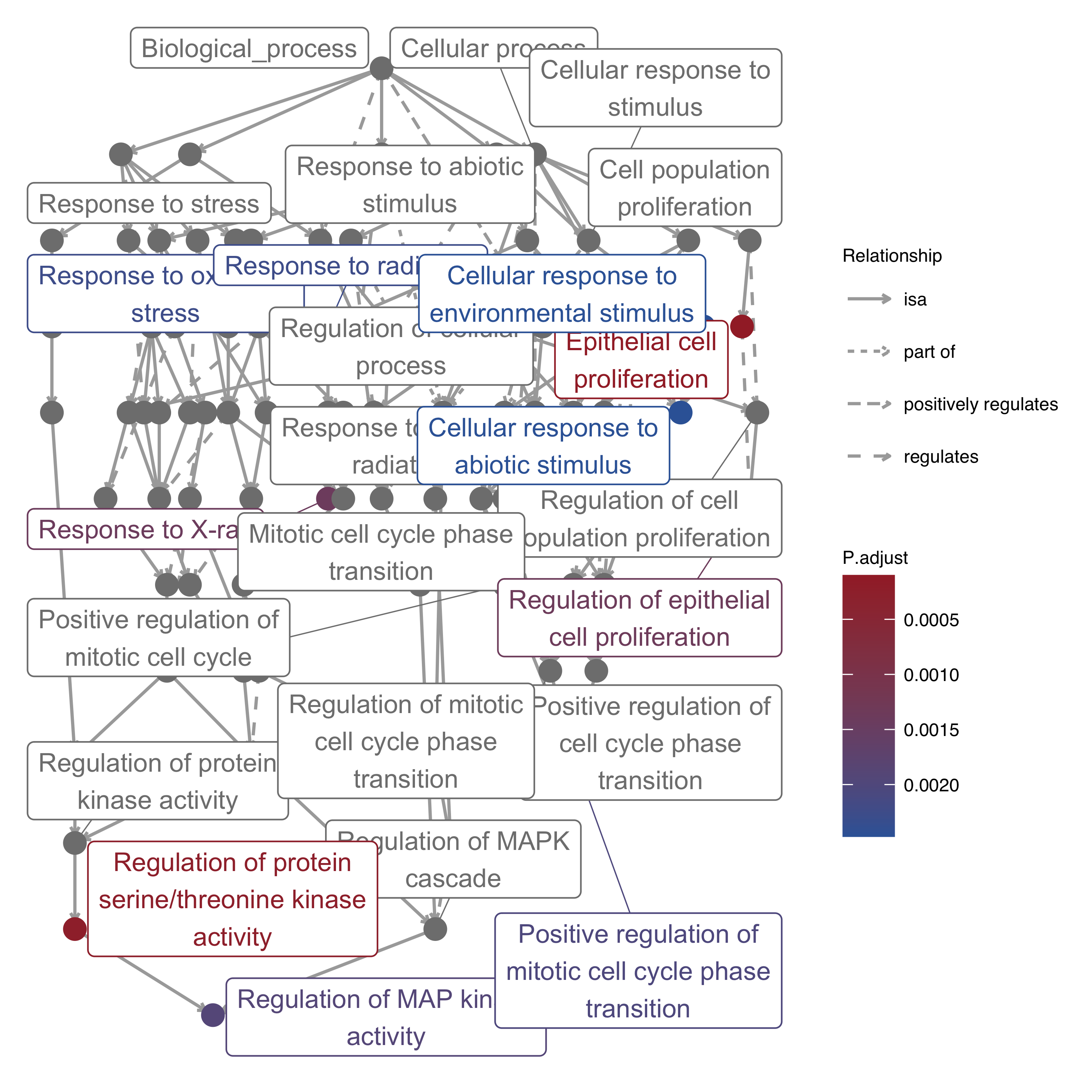
Figure 11.12: Map plot of enrichment analysis.
11.13 GO-specific: Terms Heatmap Plot
Inspired by rrvgo, genekitr borrows the main code to cluster GO terms. It can also use five methods to calculate the similarity between terms.
plotEnrich(ego, plot_type = "goheat", sim_method = "Rel")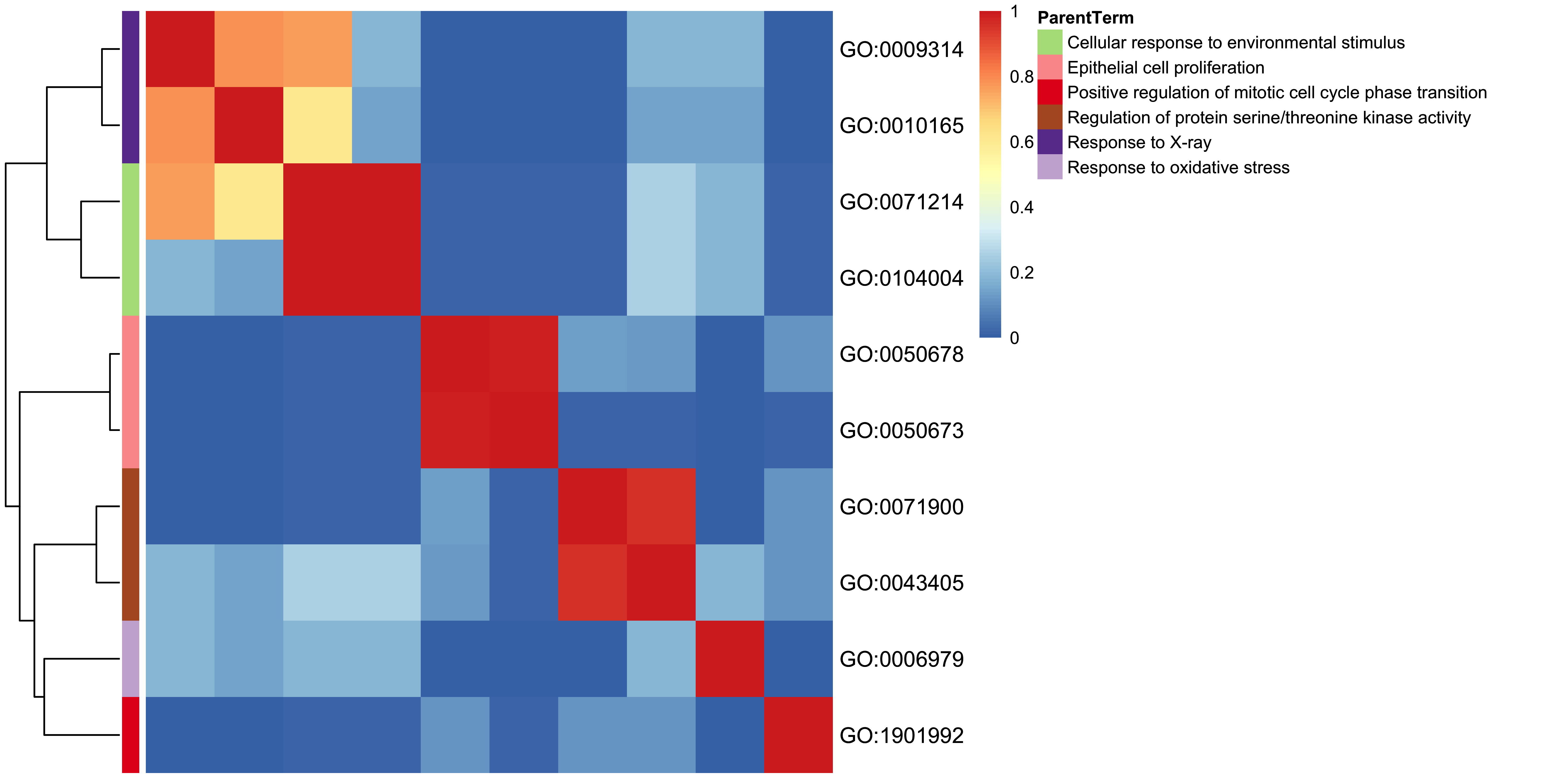
Figure 11.13: GO heatmap plot of enrichment analysis.
11.14 GO-specific: Terms Tangram Plot
According to the rrvgo vignettes, the tangram plot is a space-filling visualization of hierarchical structures. The terms are grouped and colored based on their parent, and the space used by the term is proportional to the score.
plotEnrich(ego, plot_type = "gotangram", sim_method = "Rel")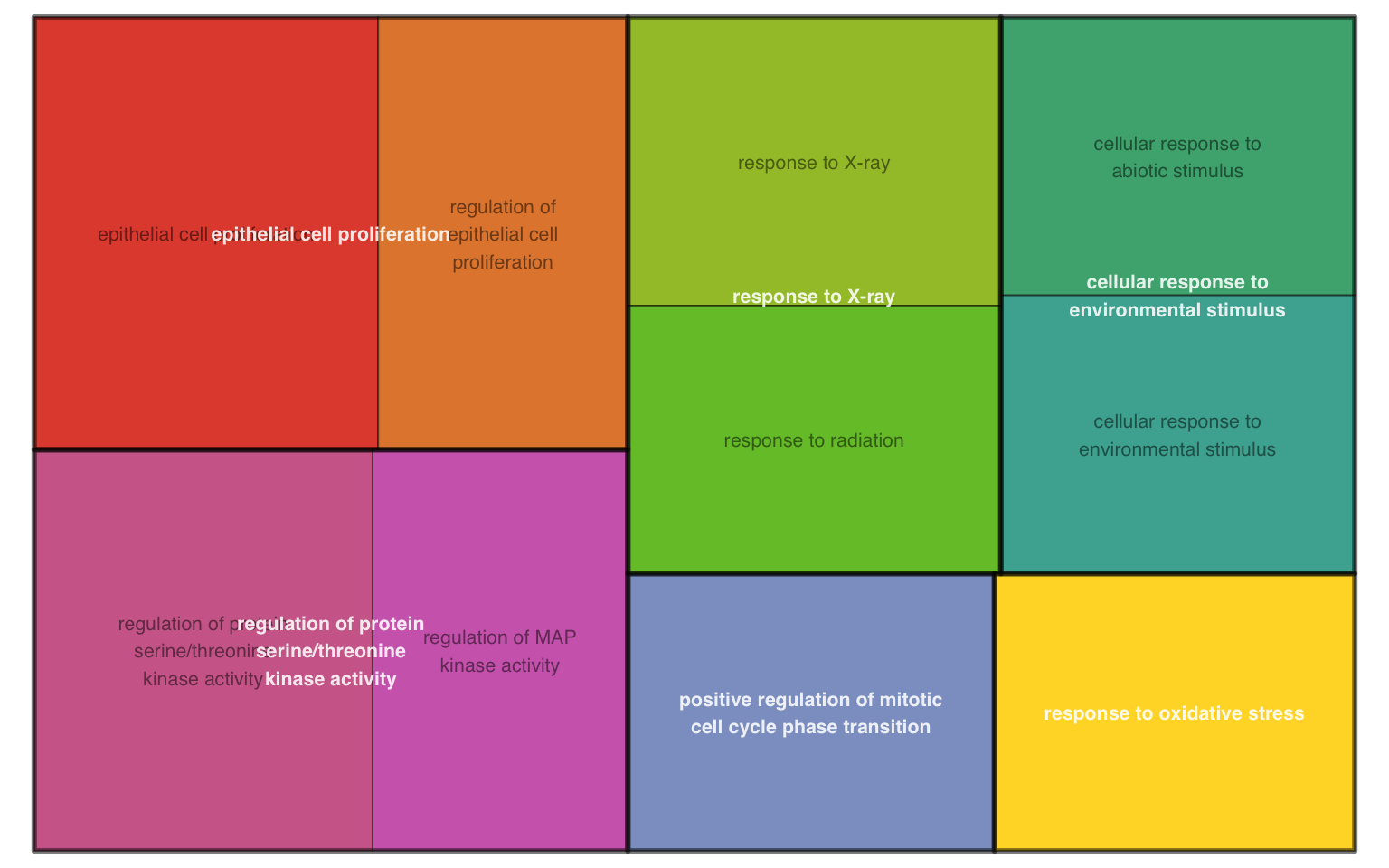
Figure 11.14: GO tangram plot of enrichment analysis.
11.15 Plot Theme
Genekitr provides a function plot_theme() to modify all plot themes, including text size, border, legend, color, etc.
library(patchwork)
p1 <- plotEnrich(ego, plot_type = "dot")
p2 <- plotEnrich(ego,
plot_type = "dot",
main_text_size = 10,
legend_text_size = 10
)
p3 <- plotEnrich(ego,
plot_type = "dot",
border_thick = 3,
remove_grid = F
)
p4 <- plotEnrich(ego,
plot_type = "dot",
remove_main_text = T,
remove_legend_text = T,
remove_legend = T
)
p1 + p2 + p3 + p4 + plot_annotation(tag_levels = "A")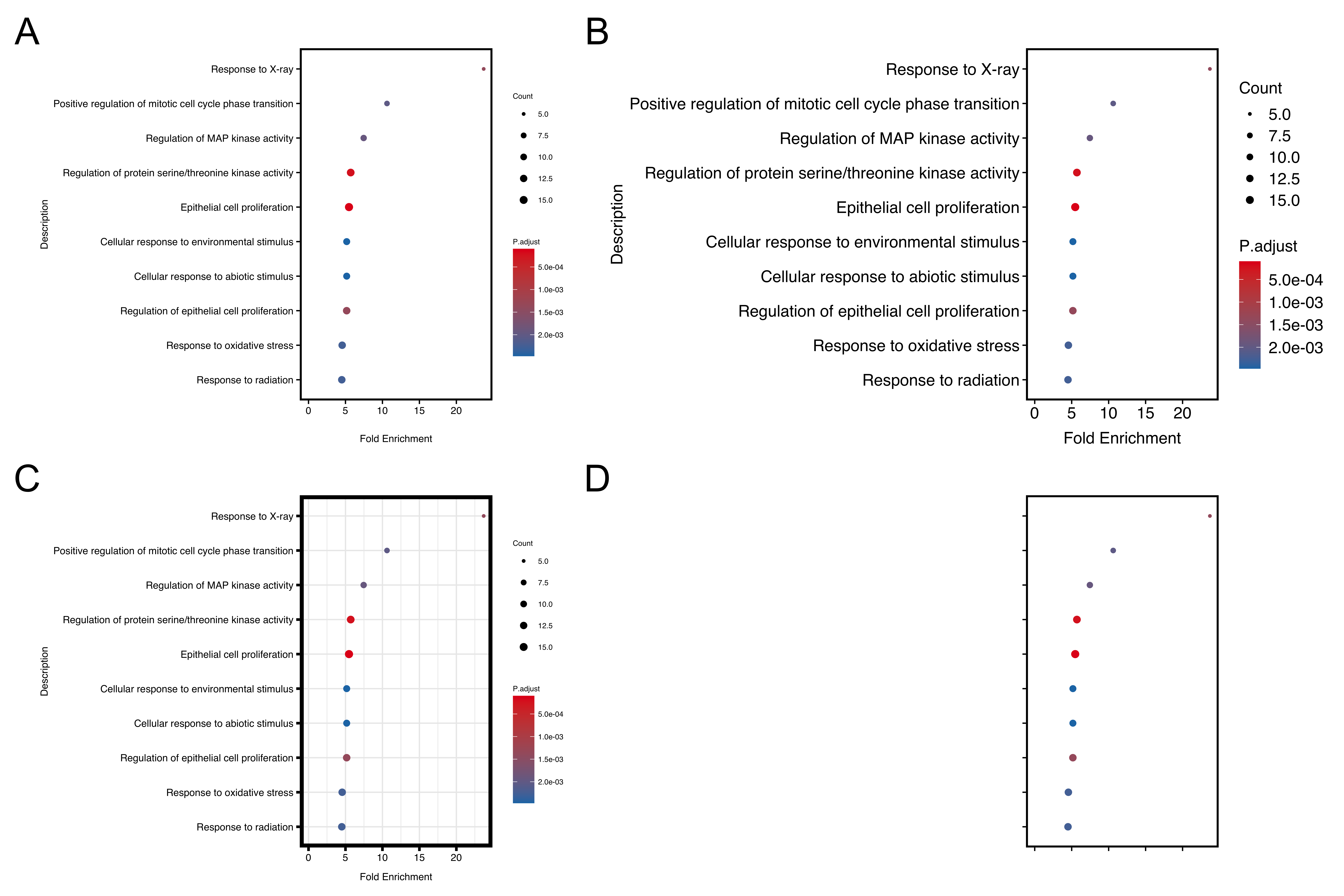
Figure 11.15: Plot theme. default theme (A), modify text size (B), modify grid line and border size (C) and remove all text and legend (D).
11.16 Advanced Plot
11.16.1 Two-group barplot for up/down regulated pathways
Up- and down-regulated genes can be passed to genORA separately and visualized together as a two-group barplot.
Here we take the GO result as an example:
# 1st step: prepare input IDs
# Since the geneList is logFC decreasing ordered, we could take first 100 as up-regulated genes and vice versa.
data(geneList, package = "genekitr")
up_genes <- head(names(geneList), 100)
down_genes <- tail(names(geneList), 100)
# 2nd step: prepare gene set
hg_gs <- geneset::getGO(org = "human",ont = "bp")
# 3rd step: ORA analysis separately
up_go <- genORA(up_genes, geneset = hg_gs)
down_go <- genORA(down_genes, geneset = hg_gs)
dim(up_go)## [1] 29 12
dim(down_go)## [1] 272 12There are two visualization types:
-
plot_type = "one": both up and down-regulated pathways are plotted together -
plot_type = "two": up and down-regulated pathways are plotted separately. Recommended if the number of both groups is similar.
plotEnrichAdv(up_go, down_go,
plot_type = "one",
term_metric = "FoldEnrich",
stats_metric = "p.adjust",
xlim_left = 25, xlim_right = 15) +
theme(legend.position = c(0.15, 0.9))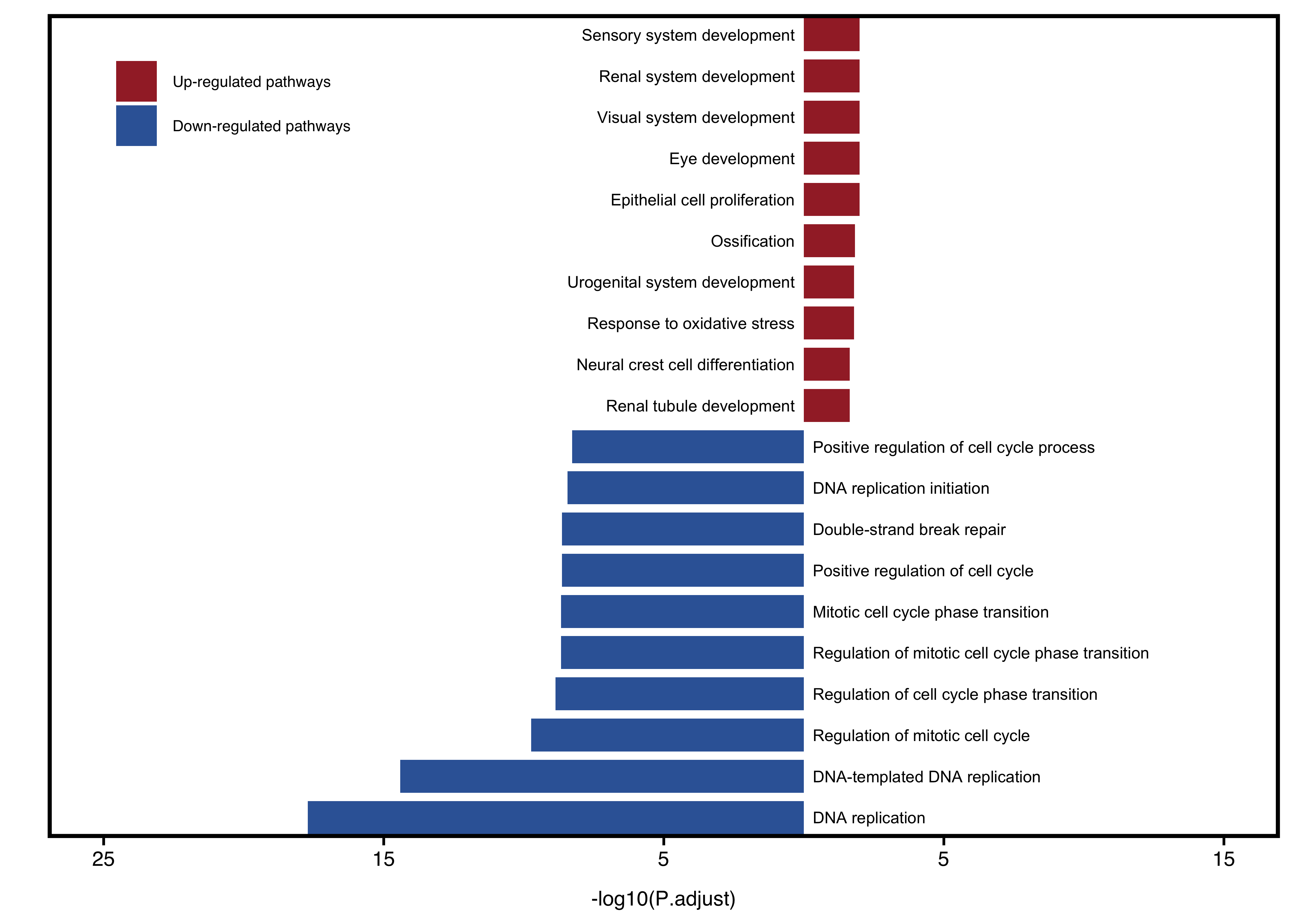
Figure 11.16: Visualize two groups together when plot_type = "one".
plotEnrichAdv(up_go, down_go,
plot_type = "two",
term_metric = "FoldEnrich",
stats_metric = "qvalue"
)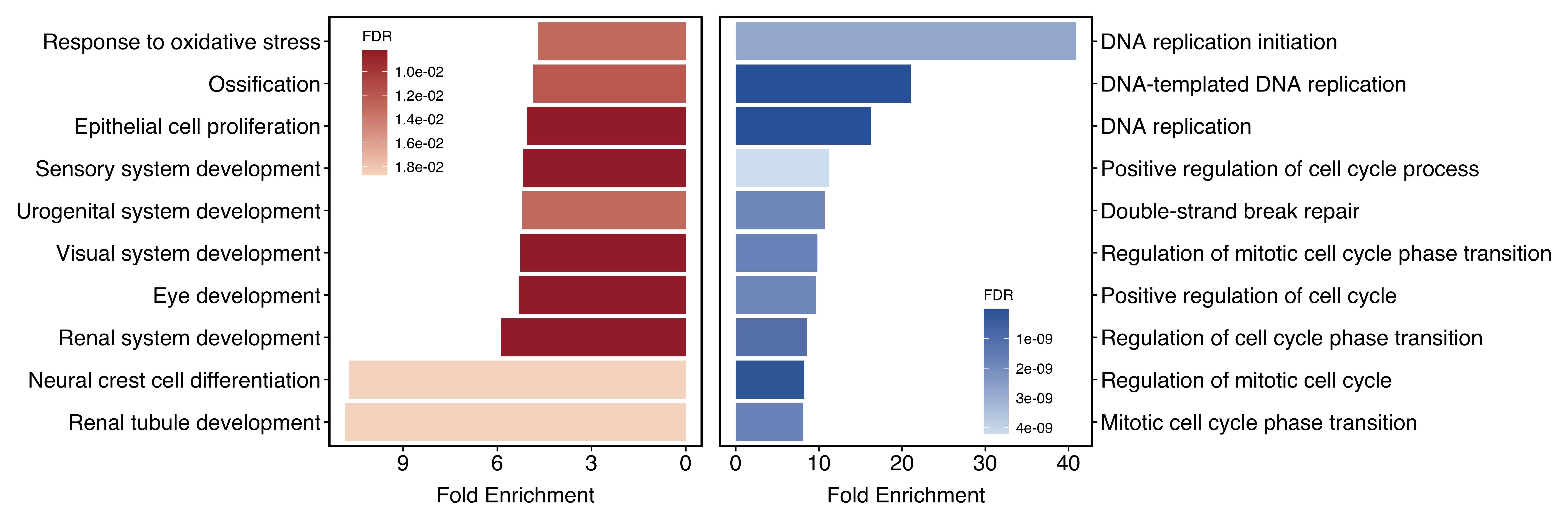
Figure 11.17: Visualize two groups separately when plot_type = "two".