12 Plot FCS-GSEA
GSEA analysis returns a list result, there are two ways of visulization:
Directly pass the list to
plotGSEAwhich enables 5 types (classic pathway plot, volcano plot, multi-pathway plot, ridge plot and two-side bar plot).Extract the analysis result (e.g.
gse_list$gsea_df) then pass toplotEnrich, which enables 8 types (geneheat, genechord, wordcloud, upset, network, gomap, goheat, gotangram).
12.1 Get GSEA result
For more details, please refer to chapter9
## 948 1638 158471 10610 6947 100133941
## 5.780170 5.633027 4.683610 3.875120 3.357670 3.32253312.2 Classic Pathway Plot
Running enrichment score of a gene set is drawn as classic pathway plot lines. The function will walks down the ranked gene list. The vertical lines in the middle show the location of gene set members.
If user wants to show specific pathways or genes, just pass to according parameters:
- show_pathway: IDs in GSEA result
- show_gene: IDs in GSEA result
pathways <- c("HALLMARK_P53_PATHWAY", "HALLMARK_GLYCOLYSIS", "HALLMARK_DNA_REPAIR")
genes <- c("MET", "TP53", "PMM2")
plotGSEA(gse, plot_type = "classic", show_pathway = pathways, show_gene = genes)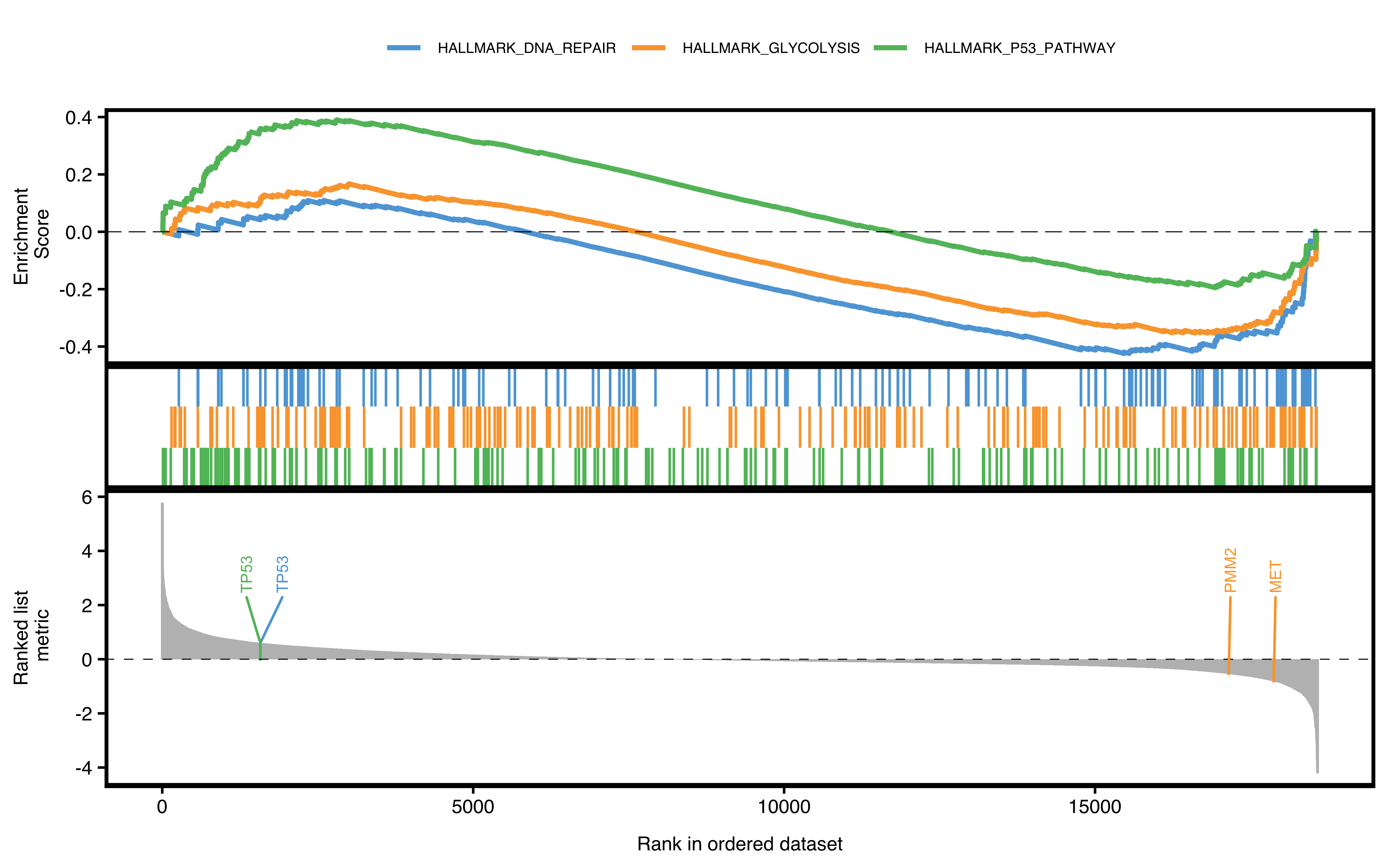
Figure 12.1: Classic pathway plot of GSEA.
12.3 Volcano Pathway Plot
In this plot, the degree of enrichment is indicated by a normalized enrichment score or NES.
Slightly different with DEG volcano plot, the x-axis is represented as NES, which corrects for enrichment score differences among gene-sets.
A significant positive NES value indicates that members of the gene set tend to appear at the top of the ranked data (e.g. fold change of DEG) and a significant negative NES indicates the opposite.
Default shows the top(N) pathways. If user wants to show specific pathway, also pass the argument to show_pathway.
library(patchwork)
pathways <- c("HALLMARK_P53_PATHWAY", "HALLMARK_GLYCOLYSIS", "HALLMARK_DNA_REPAIR")
p1 <- plotGSEA(gse, plot_type = "volcano", show_pathway = 3)
p2 <- plotGSEA(gse, plot_type = "volcano", show_pathway = pathways)
p1 + p2 + plot_annotation(tag_levels = "A")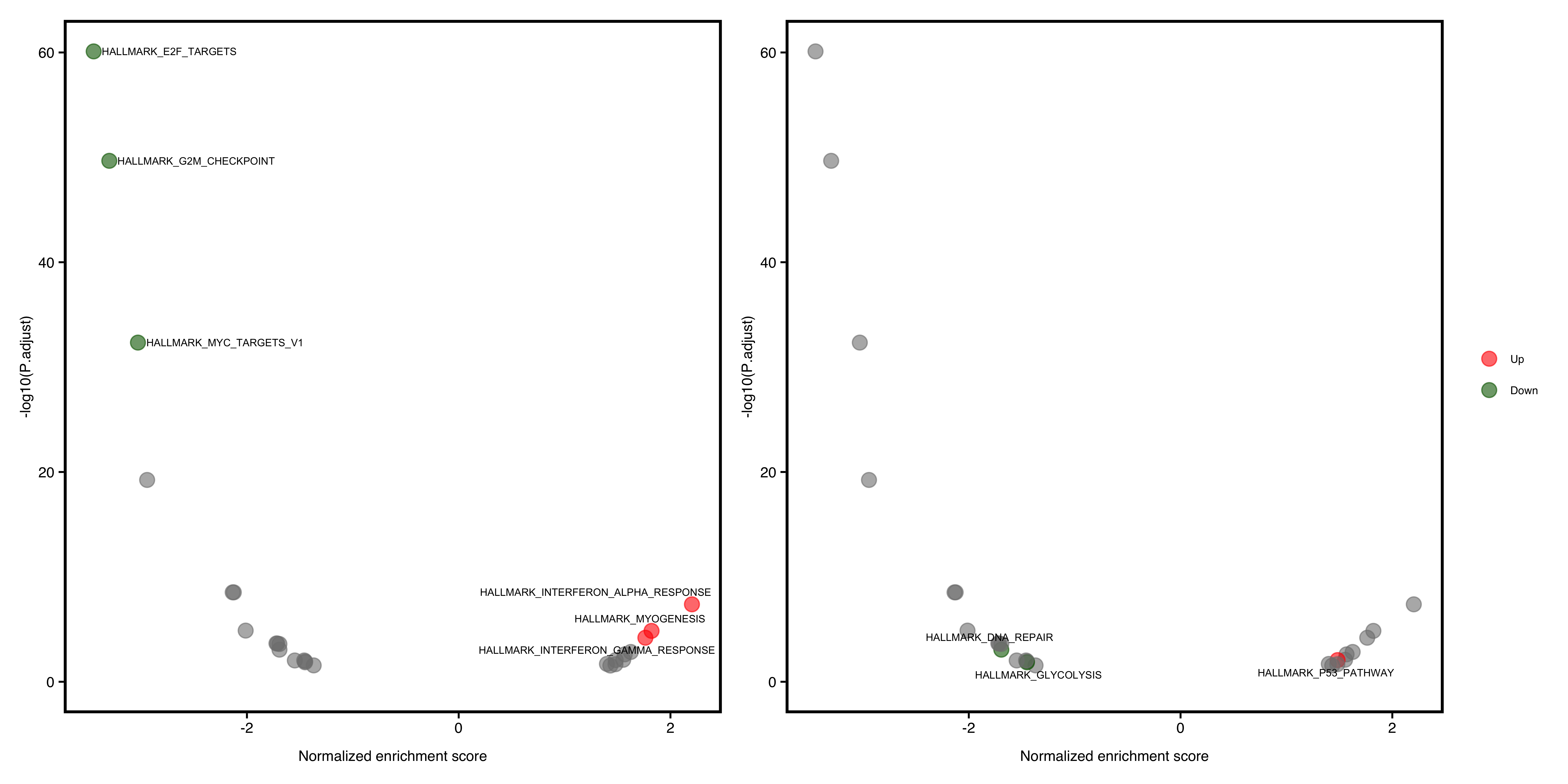
Figure 12.2: Volcano pathway plot of GSEA. default (A), select pathways (B).
12.4 Multi-pathway Plot
If user wants to show a bunch of selected pathways, plotGSEA has scratched some codes from fgsea.
Default shows the top(N) pathways. If user wants to show specific pathway, also pass the argument to show_pathway.
library(patchwork)
pathways <- c("HALLMARK_P53_PATHWAY", "HALLMARK_GLYCOLYSIS", "HALLMARK_DNA_REPAIR")
p1 <- plotGSEA(gse, plot_type = "fgsea", show_pathway = 5)
p2 <- plotGSEA(gse, plot_type = "fgsea", show_pathway = pathways)
p1 + p2 + plot_annotation(tag_levels = "A")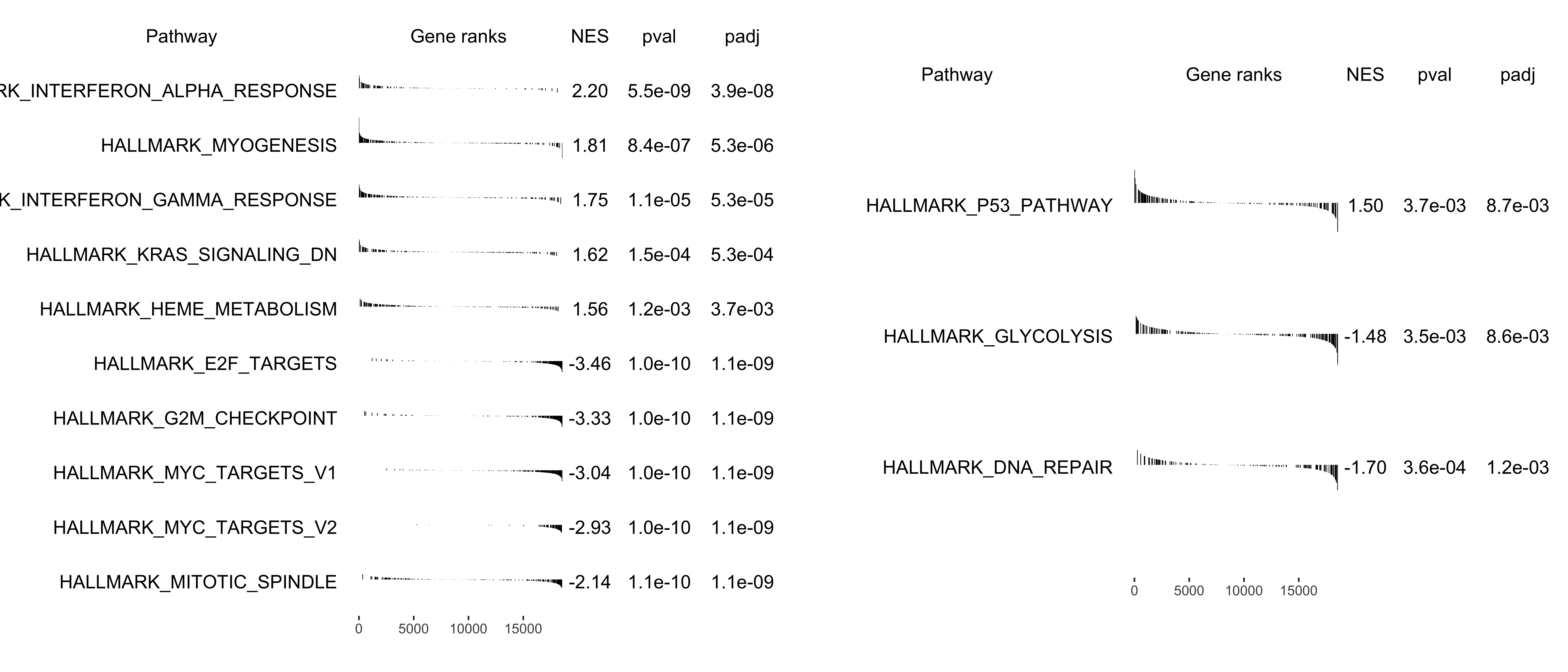
Figure 12.3: Multi-pathway plot of GSEA. default (A), select pathways (B).
12.5 Ridge Plot
Ridge plot will visualize enriched pathways of up/down regulated genes. The x-axis is fold change and y-axis is pathways. Color represents statistical value (e.g. pvalue/p.adjust/qvalue)
plotGSEA(gse,
plot_type = "ridge",
show_pathway = 10, stats_metric = "p.adjust"
)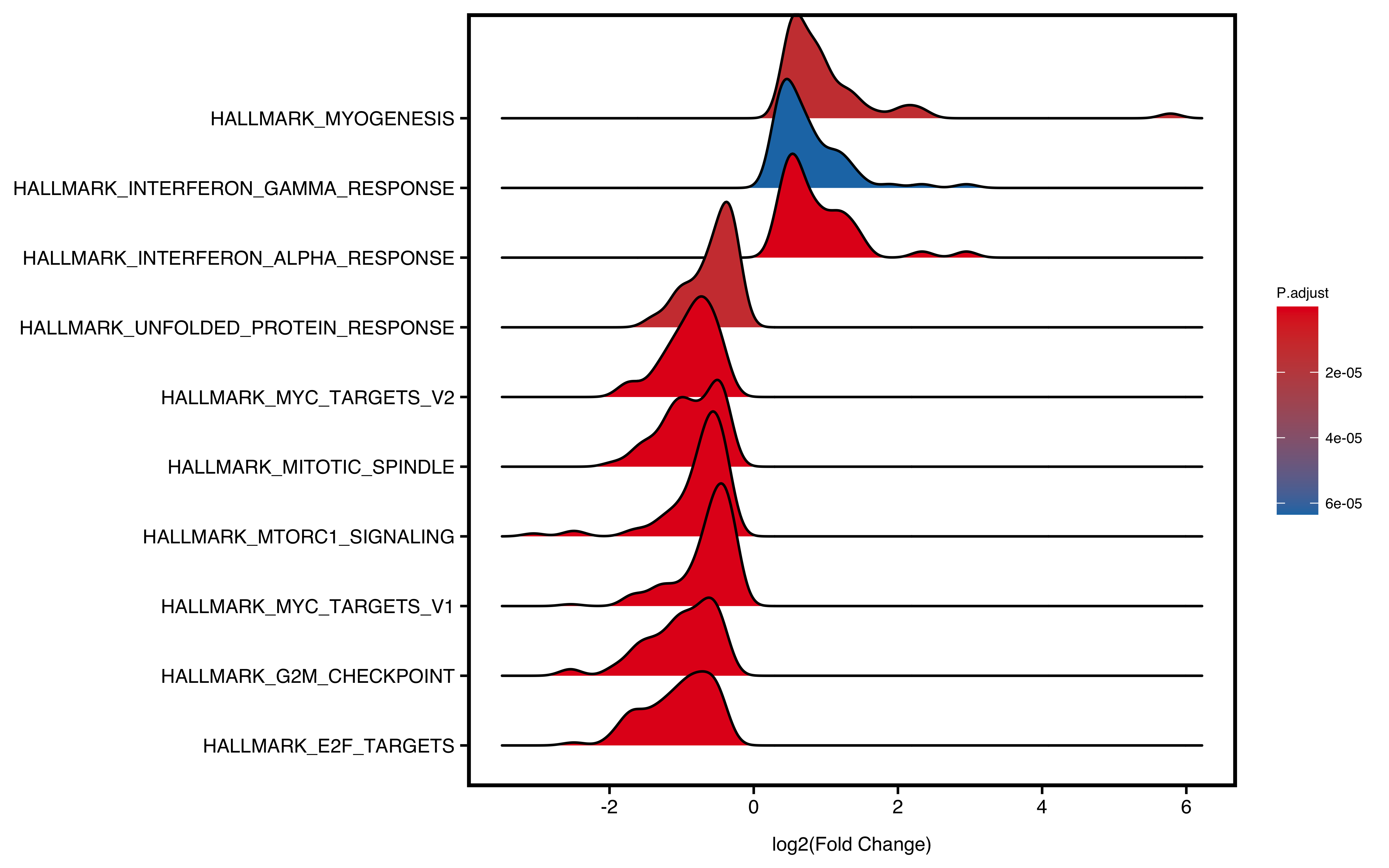
Figure 12.4: Ridge plot of GSEA.
12.6 Two-side Barplot
The barplot will separate positive and negative NES into two sides. User could specify color for the two sides. Meanwhile, non-significant pathways (e.g. Pvalue > 0.05) will be colored as “grey”.
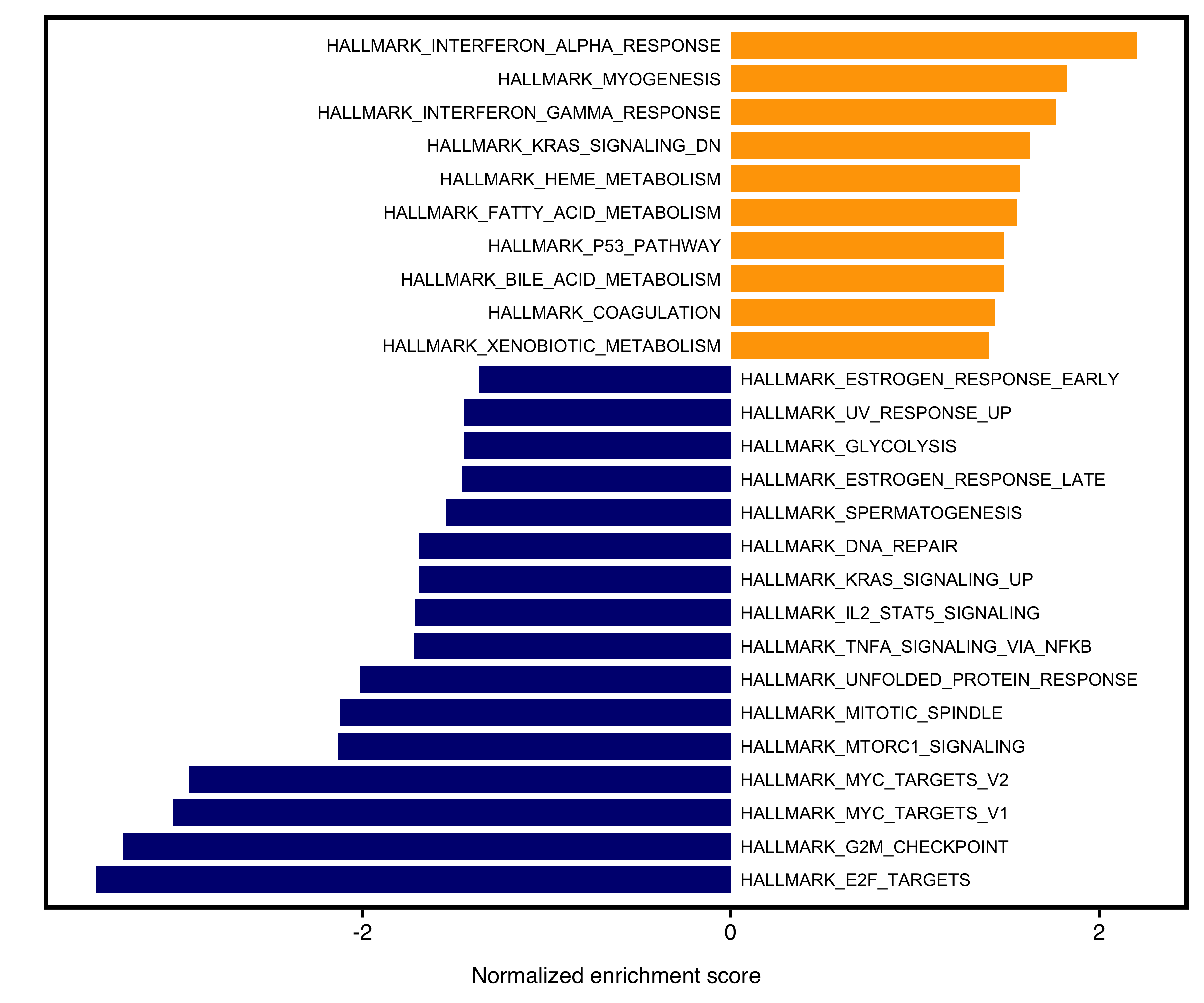
Figure 12.5: Two-side bar plot of GSEA.
12.7 Borrow from plotEnrich
More plotting details at chapter 11
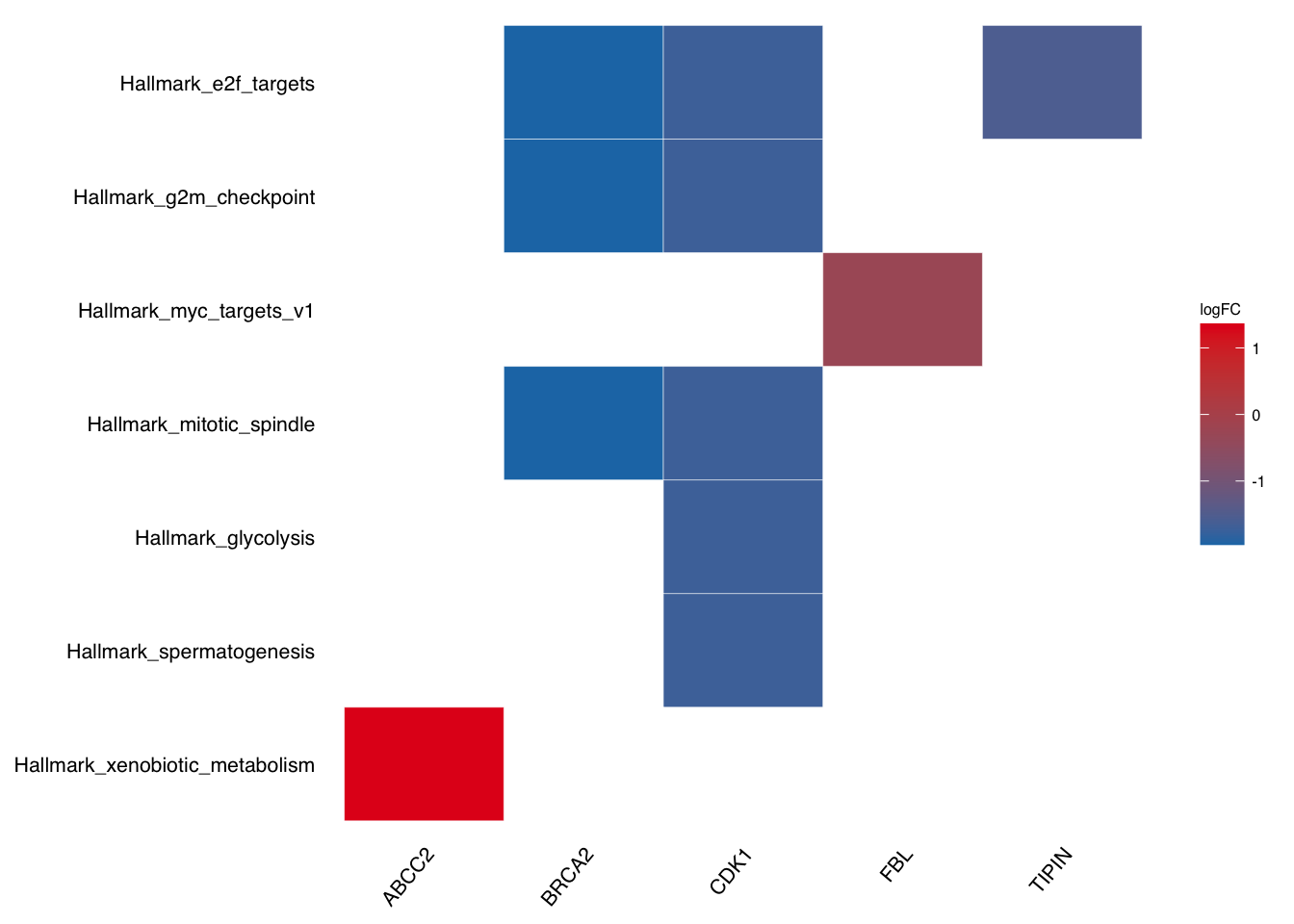
Take geneheat plot as example:
plotEnrich(gse$gsea_df,
plot_type = "geneheat",
show_gene = c("BRCA2", "CDK1", "MCM8", "TIPIN","FBL","ABCC2"),
fold_change = geneList)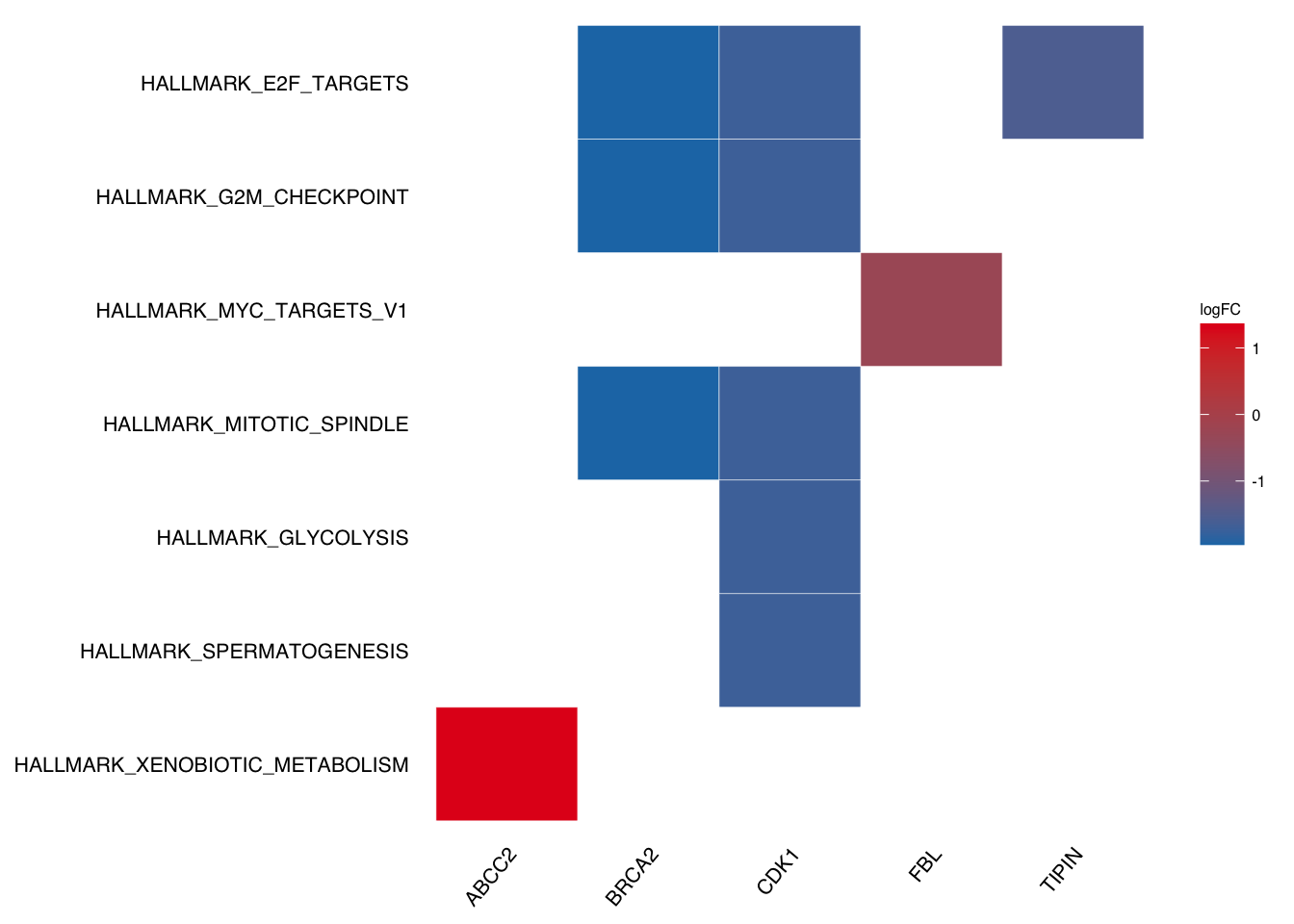
12.8 Tricks
12.8.1 Change figure labels
If we want to DIY labels (e.g. make lower case for above plot), we need to figure out which column the labels from.
In the above geneheat plot, the labels are from Description column so we only need to change the column.
gse2 <- gse
gse2$gsea_df$Description <- tolower(gse2$gsea_df$Description)
# plotEnrich will uppercase the first letter automatically.
plotEnrich(gse2$gsea_df,
plot_type = "geneheat",
show_gene = c("BRCA2", "CDK1", "MCM8", "TIPIN","FBL","ABCC2"),
fold_change = geneList)